Fenyloketonuria
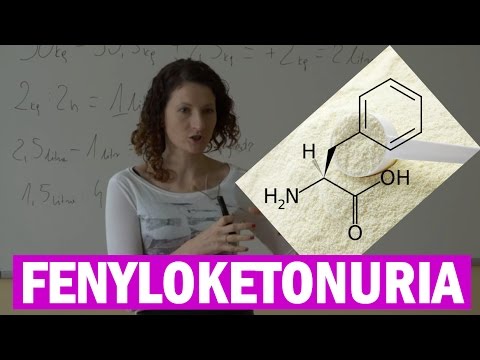
Fenyloketonuria (PKU) to rzadka choroba, w której dziecko rodzi się bez zdolności do prawidłowego rozkładu aminokwasu zwanego fenyloalaniną.
Fenyloketonuria (PKU) jest dziedziczna, co oznacza, że jest przekazywana przez rodziny. Oboje rodzice muszą przekazać niedziałającą kopię genu, aby dziecko miało tę chorobę. W takim przypadku ich dzieci mają 1 na 4 szanse na to.
U niemowląt z PKU brakuje enzymu zwanego hydroksylazą fenyloalaniny. Jest potrzebny do rozbicia niezbędnego aminokwasu fenyloalaniny. Fenyloalanina znajduje się w żywności zawierającej białko.
Bez tego enzymu w organizmie narasta poziom fenyloalaniny. To nagromadzenie może uszkodzić ośrodkowy układ nerwowy i spowodować uszkodzenie mózgu.
Fenyloalanina odgrywa rolę w produkcji melaniny przez organizm. Pigment odpowiada za kolor skóry i włosów. Dlatego niemowlęta z tą chorobą często mają jaśniejszą skórę, włosy i oczy niż bracia lub siostry bez choroby.
Inne objawy mogą obejmować:
- Opóźnione umiejętności umysłowe i społeczne
- Rozmiar głowy znacznie mniejszy niż normalnie
- Nadpobudliwość
- Szarpanie rąk lub nóg
- Niepełnosprawność umysłowa
- Napady padaczkowe
- Wysypki skórne
- Wstrząsy
Jeśli PKU nie jest leczona lub jeśli spożywane są pokarmy zawierające fenyloalaninę, oddech, skóra, woskowina i mocz mogą mieć „mysi” lub „stęchły” zapach. Ten zapach jest spowodowany gromadzeniem się substancji fenyloalaniny w organizmie.
PKU można łatwo wykryć za pomocą prostego badania krwi. Wszystkie stany w Stanach Zjednoczonych wymagają badania przesiewowego PKU dla wszystkich noworodków w ramach panelu badań przesiewowych noworodków. Test jest zwykle wykonywany przez pobranie kilku kropli krwi od dziecka, zanim dziecko opuści szpital.
Jeśli test przesiewowy jest pozytywny, wymagane są dalsze badania krwi i moczu w celu potwierdzenia diagnozy. Wykonywane są również badania genetyczne.
PKU jest chorobą uleczalną. Leczenie obejmuje dietę o bardzo niskiej zawartości fenyloalaniny, szczególnie gdy dziecko rośnie. Dieta musi być ściśle przestrzegana. Wymaga to ścisłego nadzoru dyplomowanego dietetyka lub lekarza oraz współpracy rodzica i dziecka. Ci, którzy kontynuują dietę w wieku dorosłym, mają lepsze zdrowie fizyczne i psychiczne niż ci, którzy na niej nie pozostają. „Dieta na całe życie” stała się standardem zalecanym przez większość ekspertów. Kobiety z PKU muszą przestrzegać diety przed poczęciem i przez cały okres ciąży.
W mleku, jajach i innych popularnych pokarmach znajdują się duże ilości fenyloalaniny. Sztuczny słodzik NutraSweet (aspartam) zawiera również fenyloalaninę. Należy unikać wszelkich produktów zawierających aspartam.
Istnieje kilka specjalnych preparatów dla niemowląt z PKU. Mogą być one wykorzystywane jako źródło białka o wyjątkowo niskiej zawartości fenyloalaniny i zrównoważone dla pozostałych niezbędnych aminokwasów. Starsze dzieci i dorośli stosują inną formułę, która dostarcza białka w ilościach, których potrzebują. Osoby z PKU muszą przyjmować formułę codziennie przez całe życie.
Oczekuje się, że wynik będzie bardzo dobry, jeśli dieta będzie ściśle przestrzegana, zaczynając wkrótce po urodzeniu dziecka. Jeśli leczenie jest opóźnione lub stan pozostaje nieleczony, nastąpi uszkodzenie mózgu. Funkcjonowanie w szkole może być lekko upośledzone.
Jeśli nie unika się białek zawierających fenyloalaninę, PKU może prowadzić do upośledzenia umysłowego pod koniec pierwszego roku życia.
Poważne upośledzenie umysłowe występuje, jeśli zaburzenie nie jest leczone. ADHD (zespół nadpobudliwości psychoruchowej z deficytem uwagi) wydaje się być częstym problemem u tych, którzy nie trzymają się diety o bardzo niskiej zawartości fenyloalaniny.
Zadzwoń do swojego lekarza, jeśli Twoje dziecko nie zostało przebadane na PKU. Jest to szczególnie ważne, jeśli ktoś z Twojej rodziny cierpi na tę chorobę.
Test enzymatyczny lub testy genetyczne mogą określić, czy rodzice są nosicielami genu PKU. Podczas ciąży można wykonać biopsję kosmówki lub amniopunkcję w celu zbadania nienarodzonego dziecka na obecność PKU.
Bardzo ważne jest, aby kobiety z PKU ściśle przestrzegały ścisłej diety niskofenyloalaniny zarówno przed zajściem w ciążę, jak i przez cały okres ciąży. Nagromadzenie fenyloalaniny uszkodzi rozwijające się dziecko, nawet jeśli dziecko nie odziedziczyło pełnej choroby.
PKU; Fenyloketonuria noworodkowa
 Test fenyloketonurii
Test fenyloketonurii Badania przesiewowe noworodków
Badania przesiewowe noworodków
Kliegman RM, St. Geme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM. Wady metabolizmu aminokwasów. W: Kliegman RM, St. Geme JW, Schor NF, Blum NJ, Shah SS, Tasker RC, Wilson KM, wyd. Podręcznik Pediatrii Nelsona. Wyd. 21. Filadelfia, Pensylwania: Elsevier; 2020:rozdz. 103.
Kumar V, Abbas AK, Aster JC. Choroby genetyczne i pediatryczne. W: Kumar V, Abbas AK, Aster JC, wyd. Podstawowa patologia rudzików. 10. wyd. Filadelfia, Pensylwania: Elsevier; 2018:rozdz. 7.
Vockley J, Andersson HC, Antshel KM, et al; American College of Medical Genetics and Genomics Therapeutics Committee. Niedobór hydroksylazy fenyloalaniny: wytyczne dotyczące diagnostyki i postępowania. Genet Med. 2014;16(2):188-200. PMID: 24385074 www.ncbi.nlm.nih.gov/pubmed/24385074.
Polecamy Cię

Ryby suajskie: jeść czy unikać?
